臨床試驗是證明產(chǎn)品安全性有效性的重要途徑和手段���。醫(yī)療器械臨床試驗是指在相應(yīng)的臨床環(huán)境中,對擬申請注冊的普通醫(yī)療器械在正常使用條件下的安全性和有效性進行確認或者驗證的過程���,或?qū)w外診斷試劑的臨床性能進行系統(tǒng)性研究的過程�。
很多朋友經(jīng)常來問�����,我這個試驗產(chǎn)品準(zhǔn)備開展醫(yī)療器械臨床試驗����,做多少例受試者合適,能不能找一個最低的例數(shù)��,這樣的話,整個研發(fā)成本會比較低�����。例數(shù)太多�,經(jīng)費太高,預(yù)算也不好做����,老板不同意。而例數(shù)太少����,審評通不過���,整個試驗在注冊時���,會面臨退審或者發(fā)補。但是開展多少例���,NMPA也沒有給一個明確的例數(shù)�����。
療器械臨床試驗.jpg")
對于一些把項目委托出來的公司�,在面臨不同CRO報價時,得到的例數(shù)也不盡相同��,然后就開始茫然了�����。
對于一個試驗究竟需要開展多少例�����?不是盲從以往項目的經(jīng)驗����,也不是隨隨便便就開展,還是從科學(xué)的角度�,進行設(shè)計和計算。具體還是要重視統(tǒng)計專家���。
一�、CFDA對于醫(yī)療器械病例數(shù)的要求(不含IVD)
1.1《醫(yī)療器械臨床試驗質(zhì)量管理規(guī)范》
第二十七條未在境內(nèi)外批準(zhǔn)上市的新產(chǎn)品����,安全性以及性能尚未經(jīng)醫(yī)學(xué)證實的�����,臨床試驗方案設(shè)計時應(yīng)當(dāng)先進行小樣本可行性試驗�,待初步確認其安全性后����,再根據(jù)統(tǒng)計學(xué)要求確定樣本量開展后續(xù)臨床試驗。
1.2《產(chǎn)品注冊指導(dǎo)原則》
對于有指導(dǎo)原則的產(chǎn)品�,可以參考指導(dǎo)原則。雖然有指導(dǎo)原則��,具體還是要根據(jù)產(chǎn)品的特點���、特性進行評估。
例如:某指導(dǎo)原則對于樣本量要求:樣本量根據(jù)受試產(chǎn)品的臨床試驗設(shè)計類型����、主要評價指標(biāo)等因素來確定。需詳細寫明樣本量估算采用的軟件或公式�,以及公式中的所有參數(shù)及其估計值,還應(yīng)結(jié)合臨床實際情況考慮試驗對象的可能脫落率等因素�。對于非劣效試驗設(shè)計,應(yīng)由臨床專家和統(tǒng)計學(xué)家事先給出具有臨床意義的非劣效界值��。對于單組目標(biāo)值設(shè)計,亦需明確給出目標(biāo)值確定的合理依據(jù)���。
所以具體多少例還是要根據(jù)產(chǎn)品特點����、設(shè)計類型���,設(shè)計方法�����、主要療效終點多方位因素進行統(tǒng)計計算�����。
二��、歷年注冊NMPA提出的醫(yī)療器械方案病例數(shù)的問題
2.1 未提供樣本量的具體計算過程及確定依據(jù)��。
2.2未提供樣本量計算公式中各參數(shù)的確定依據(jù),如:非劣效界值���。
2.3未提供臨床隨訪時間的確定依據(jù)。
2.4未明確主要評價指標(biāo)、次要評價指標(biāo)及其相關(guān)依據(jù)���。
2.5未明確主要終點指標(biāo)及時間窗的選擇依據(jù)���。
可見NMPA對于病例數(shù)還是很重視的,千萬要重視��。
三����、如何開展一項臨床試驗,并保證病例數(shù)合適
3.1《醫(yī)療器械臨床試驗設(shè)計指導(dǎo)原則》
臨床試驗收集受試人群中的療效/安全性數(shù)據(jù)���,用統(tǒng)計分析將基于主要評價指標(biāo)的試驗結(jié)論推斷到與受試人群具有相同特征的目標(biāo)人群�。為實現(xiàn)樣本(受試人群)代替總體(目標(biāo)人群)的目的��,臨床試驗需要一定的受試者數(shù)量(樣本量)���。樣本量大小與主要評價指標(biāo)的變異度呈正相關(guān),與主要評價指標(biāo)的組間差異呈負相關(guān)�。
樣本量一般以臨床試驗的主要評價指標(biāo)進行估算。需在臨床試驗方案中說明樣本量估算的相關(guān)要素及其確定依據(jù)����、樣本量的具體計算方法�����。后文提供了樣本量估算公式的樣例�����,供參考���。確定樣本量的相關(guān)要素一般包括臨床試驗的設(shè)計類型和比較類型、主要評價指標(biāo)的類型和定義���、主要評價指標(biāo)有臨床實際意義的界值�、主要評價指標(biāo)的相關(guān)參數(shù)(如預(yù)期有效率���、均值��、標(biāo)準(zhǔn)差等)����、Ⅰ類和Ⅱ類錯誤率以及預(yù)期的受試者脫落和方案違背的比例等�����。主要評價指標(biāo)的相關(guān)參數(shù)根據(jù)已有臨床數(shù)據(jù)和小樣本可行性試驗(如有)的結(jié)果來估算,需要在臨床試驗方案中明確這些估計值的確定依據(jù)���。一般情況下���,Ⅰ類錯誤概率α設(shè)定為雙側(cè)0.05或單側(cè)0.025,Ⅱ類錯誤概率β設(shè)定為不大于0.2�,預(yù)期受試者脫落和方案違背的比例不大于0.2,申請人可根據(jù)產(chǎn)品特征和試驗設(shè)計的具體情形采用不同的取值�����,需充分論證其合理性�����。
3.2計算公式
決定樣本量的關(guān)鍵因素有:產(chǎn)品特點��、研究類型�����、主要評價指標(biāo)�����、對照組與試驗組主要評價指標(biāo)的預(yù)期療效���、非劣效界值或目標(biāo)值��、顯著性水平(α)��、把握度(β)����、預(yù)期失訪率等�。
3.2.1 平行對照設(shè)計樣本量估算
以下公式中,nT�、nC分別為試驗組和對照組的樣本量;Z1-α/2�����、Z1-β為標(biāo)準(zhǔn)正態(tài)分布的分數(shù)位��,當(dāng)α=0.05時�����,Z1-α/2=1.96,當(dāng)β=0.2時�,Z1-β=0.842;(Z1-α/2+Z1-β)2=7.85
(一)優(yōu)效性試驗
當(dāng)試驗組和對照組按照1:1隨機化分組����,主要評價指標(biāo)為事件發(fā)生率,其方差齊且不接近于0%或100%時��,其樣本量估算公式為:
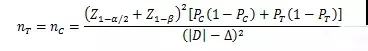
PT���、PC分別為試驗組和對照組預(yù)期事件發(fā)生率����;為兩組預(yù)期率差的絕對值��, = ���;Δ為優(yōu)效性界值��,取正值����。
試驗組和對照組按照1:1隨機化分組�����,主要評價指標(biāo)為定量指標(biāo)且方差齊時,其樣本量估算公式為:
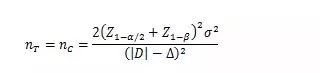
σ為對照組預(yù)期標(biāo)準(zhǔn)差�;為預(yù)期的兩組均數(shù)之差的絕對值��, = �����;Δ為優(yōu)效性界值�,取正值。
使用該公式計算樣本量為Z值計算的結(jié)果��,小樣本時宜使用t值迭代�,或總例數(shù)增加2—3例。
(二)等效性試驗
當(dāng)試驗組和對照組按照1:1隨機化分組�����,主要評價指標(biāo)為事件發(fā)生率�����,其方差齊且不接近于0%或100%時����,其樣本量估算公式為:
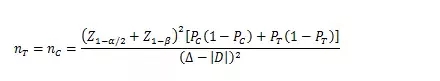
PT���、PC分別為試驗組和對照組預(yù)期事件發(fā)生率;為兩組預(yù)期率差的絕對值��, = �����;?為等效界值(適用于劣側(cè)界值與優(yōu)側(cè)界值相等的情形)�����,取正值�����。
當(dāng)試驗組和對照組按照1:1隨機化分組�,主要評價指標(biāo)為定量指標(biāo)且方差齊時,其樣本量估算公式為:
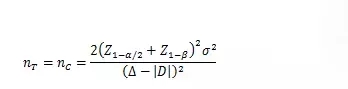
σ為對照組預(yù)期標(biāo)準(zhǔn)差�;為預(yù)期的兩組均數(shù)之差的絕對值, = ��;?為等效界值(適用于劣側(cè)界值與優(yōu)側(cè)界值相等的情形),取正值���。
使用該公式計算樣本量為Z值計算的結(jié)果�,小樣本時宜使用t值迭代����,或總例數(shù)增加2—3例。
(三)非劣效試驗
當(dāng)試驗組和對照組按照1:1隨機化分組��,主要評價指標(biāo)為預(yù)期事件發(fā)生率�����,其方差齊且不接近于0%或100%時����,其樣本量估算公式為:
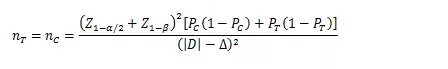
PT�、PC分別為試驗組和對照組預(yù)期事件發(fā)生率;為兩組預(yù)期率差的絕對值����, = ,?為非劣效界值�����,取負值。
當(dāng)試驗組和對照組按照1:1隨機化分組�,主要評價指標(biāo)為定量指標(biāo)且方差齊時,其樣本量估算公式為:
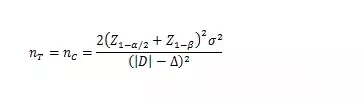
σ為對照組預(yù)期標(biāo)準(zhǔn)差�;為預(yù)期的兩組均數(shù)之差的絕對值, = ���;?為非劣效界值�����,取負值�。
使用該公式計算樣本量為Z值計算的結(jié)果��,小樣本時宜使用t值迭代�����,或總例數(shù)增加2—3例�����。
3.2.2��、單組目標(biāo)值試驗的樣本量估算
以下公式中,n為試驗組樣本量�;Z1-α/2、Z1-β為標(biāo)準(zhǔn)正態(tài)分布的分數(shù)位����,當(dāng)α=0.05時,Z1-α/2=1.96����,當(dāng)β=0.2時,Z1-β=0.842�����。
當(dāng)主要評價指標(biāo)為事件發(fā)生率��,統(tǒng)計發(fā)生率的研究周期相同��,且發(fā)生率不接近于0%或100%時�����,其樣本量估算公式為:
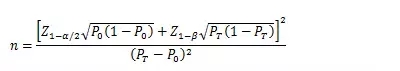
PT為試驗組預(yù)期事件發(fā)生率�,P0為目標(biāo)值�。
3.3病例數(shù)計算實例(僅供參考,具體產(chǎn)品請專業(yè)統(tǒng)計專家)
3.3.1質(zhì)子碳離子治療系統(tǒng)
假設(shè)試驗組預(yù)期有效率為95%,目標(biāo)值設(shè)為80%���,雙側(cè)顯著性水平為0.05��,把握度80%�����,根據(jù)統(tǒng)計學(xué)計算�,需要入組42例受試者���?�?紤]10%脫落�,共需要47例受試者�����。
若CTCAE 3級急性毒性反應(yīng)的比例超過5%��、或出現(xiàn)4級���、5級急性毒性反應(yīng)��,臨床試驗失敗����。
3.3.2常規(guī)牙科樹脂類充填材料

3.3.3血液透析濃縮物
非劣效試驗設(shè)計時假設(shè)對照產(chǎn)品透析達標(biāo)率為98%,預(yù)計試驗產(chǎn)品與對照產(chǎn)品的透析達標(biāo)率相當(dāng)���,臨床認可的非劣效界值為5%���,則在顯著性水平0.05(雙側(cè))、80%把握度���、考慮5%脫落率的情況下�,每組需要130例試驗對象���,兩組共需要260例試驗對象�����。
3.3.4醫(yī)用磁共振成像系統(tǒng)
根據(jù)臨床要求,影像質(zhì)量的臨床診斷優(yōu)良率不得低于75%(目標(biāo)值)(考慮到MR的圖像受患者配合的影響較大���,因此目標(biāo)值定為75%), 假設(shè)試驗組影像質(zhì)量的優(yōu)良率為90%��,則當(dāng)顯著性水平?����。p側(cè))0.05�、檢驗效能80%、考慮10%脫落率����,按統(tǒng)計學(xué)原則計算得到,試驗中每一部位最少需要的受試者數(shù)為60例���。所對應(yīng)的樣本量計算公式為:
公式中的對應(yīng)試驗組的預(yù)期療效水平�,則對應(yīng)目標(biāo)值水平����,代表標(biāo)準(zhǔn)正態(tài)分布對應(yīng)的分位數(shù),對應(yīng)統(tǒng)計檢驗的一類錯誤水平����,在此取0.025,而對應(yīng)檢驗的二類錯誤水平����,計算時取0.2�。
3.3.5一次性使用膜式氧合器
非劣效試驗設(shè)計時����,在顯著性水平0.05(雙側(cè))、80%把握度的情況下����,預(yù)期達標(biāo)率估計值95%,非劣效界值為10%����,考慮5%脫落率時,每組需要80例�,兩組合計應(yīng)至少入組160例患者。
單組目標(biāo)值設(shè)計時�,在顯著性水平0.05(雙側(cè))、80%把握度的情況下����,假設(shè)目標(biāo)值應(yīng)至少為90%,預(yù)期達標(biāo)率為95%時����,考慮5%脫落率��,試驗共需入組252例患者。
3.3.6 X射線計算機體層攝影設(shè)備
臨床試驗采用目標(biāo)值法的單組試驗����。根據(jù)臨床要求,臨床影像質(zhì)量優(yōu)良率不得低于85%(目標(biāo)值), 假設(shè)試驗組臨床影像質(zhì)量優(yōu)良率為95%�,則當(dāng)雙側(cè)顯著性水平取0.05、檢驗效能為80%�,試驗最少需要的受試者數(shù)為80例,考慮5%的脫落率�,每個部位需納入的試驗例數(shù)為不低于86例,頭頸部�����、胸部�、腹部、骨與關(guān)節(jié)四個大部位總計不低于344例���;如果預(yù)期用途中具有冠脈的部位����,冠脈需納入的試驗例數(shù)也應(yīng)不低于86例����;加上冠脈后����,所有部位總計不低于430例����。
受試者臨床試驗的部位劃分為五個,分別為:頭頸部�、胸部、腹部�����、骨與關(guān)節(jié)����、以及冠脈。冠脈掃描全部為增強掃描���,病例數(shù)不低于86例�����;除冠脈外��,增強掃描(含普通增強和血管增強)總病例數(shù)不低于80例�����,每個部位的子部位不低于5例�����。
所對應(yīng)的樣本量計算公式為:
公式中的對應(yīng)試驗組的預(yù)期療效水平����,則對應(yīng)目標(biāo)值水平����,代表標(biāo)準(zhǔn)正態(tài)分布對應(yīng)的分位數(shù),對應(yīng)統(tǒng)計檢驗的一類錯誤水平�����,在此取0.025��,而對應(yīng)檢驗的二類錯誤水平����,計算時取0.2。
3.3.7醫(yī)用X線診斷設(shè)備
根據(jù)臨床要求,影像質(zhì)量的臨床診斷要求符合率不得低于85%(目標(biāo)值), 假設(shè)試驗組影像質(zhì)量的臨床診斷要求符合率為95%��,則當(dāng)雙側(cè)顯著性水平取0.05�����、檢驗效能為80%時�,試驗最少需要的受試者數(shù)為80例。
3.3.8脊柱后路內(nèi)固定系統(tǒng)
若進行隨機對照非劣效試驗���,則需明確對照產(chǎn)品預(yù)期療效和臨床認可的非劣效界值��;申請人應(yīng)根據(jù)各自產(chǎn)品的性能指標(biāo)選擇對照品����,并采用經(jīng)典的統(tǒng)計學(xué)方法及國內(nèi)外公認的統(tǒng)計學(xué)軟件計算樣本量���。例如:假設(shè)某隨機對照非劣效臨床試驗���,根據(jù)文獻報道:其對照品的有效率為95%、臨床認可的非劣效界值為10%���,則在雙側(cè)顯著性水平0.05�����、把握度80%���、脫落率20%時��,每組需要89例。
若進行單組目標(biāo)值試驗����,則需明確試驗產(chǎn)品預(yù)期療效和臨床認可的目標(biāo)值。申請人應(yīng)提供樣本量足以評價該類產(chǎn)品安全性和有效性的統(tǒng)計學(xué)依據(jù)�����,包括以下內(nèi)容:同類產(chǎn)品臨床認可的主要評價指標(biāo)的目標(biāo)值��、受試產(chǎn)品主要評價指標(biāo)的預(yù)期療效�、I型誤差α、Ⅱ型誤差β��;所用到的樣本量計算公式�����;失訪率的合理估計;使用的統(tǒng)計軟件����;引用的參考文獻等。例如:行業(yè)認可的該類產(chǎn)品的目標(biāo)值為85%����,當(dāng)雙側(cè)顯著性水平α取0.05,β取0.2���,按照經(jīng)典的統(tǒng)計學(xué)公式���,若申報產(chǎn)品術(shù)后六個月的預(yù)期療效假設(shè)為95%,納入臨床試驗的受試者病例數(shù)至少為75例��,假設(shè)20%的失訪率�,則受試者病例數(shù)至少為90例。
3.3.9金屬接骨板內(nèi)固定系統(tǒng)
行業(yè)認可的該類產(chǎn)品有效率的目標(biāo)值為80%�,當(dāng)雙側(cè)α取0.05,β取0.2�����,按照經(jīng)典的統(tǒng)計學(xué)公式����,若申報產(chǎn)品術(shù)后6個月的預(yù)期有效率假設(shè)為95%�����,納入臨床試驗的受試者病例數(shù)至少為42例�,假設(shè)20%的失訪率�����,則受試者病例數(shù)至少為53例���;若申報產(chǎn)品術(shù)后六個月的預(yù)期有效率假設(shè)為90%,納入臨床試驗的受試者病例數(shù)至少為108例�����,假設(shè)20%的失訪率����,則受試者病例數(shù)至少為135例。
3.3.10髖關(guān)節(jié)假體系統(tǒng)
假設(shè)某隨機對照非劣效臨床試驗�����,根據(jù)文獻報道:同類產(chǎn)品的優(yōu)良率為95%、臨床認可的非劣效界值為10%��,則在雙側(cè)顯著性水平0.05��、把握度80%���、脫落率10%時�,每組需要84例��。
該研究為隨機對照非劣效臨床試驗�,主要評價指標(biāo)是術(shù)后12個月Harris評分。根據(jù)文獻報道����,對照產(chǎn)品的評分為90±10分,臨床認可的非劣效界值為5���,則在雙側(cè)顯著性水平0.05���、把握度80%、脫落率10%時����,每組需70例���。
該研究為單組目標(biāo)值試驗,主要評價指標(biāo)是術(shù)后12個月Harris評分“優(yōu)良率”�。根據(jù)文獻報道,研究產(chǎn)品的優(yōu)良率為95%����,臨床認可的目標(biāo)值為85%,則在雙側(cè)顯著性水平0.05�����、把握度80%����、脫落率10%時�����,需87例���。
3.3.11人工頸椎間盤假體
假設(shè)某隨機對照非劣效臨床試驗���,根據(jù)文獻報道:同類產(chǎn)品的治療成功率為95%���、臨床認可的非劣效界值為10%,則在雙側(cè)顯著性水平0.05��、把握度80%�����、須每組完成有效病例74例���,考慮脫落率20%時�,每組需要89例�。